
Shistocytes indicate intravascular hemolysis

Disseminated Intravascular Coagulation (DIC):
Characteristics:
– Activation of coagulation
– Generation of thrombin
– Consumption of clotting factors
– Destruction of platelets
Lab findings:
-Elevated PT (due to consumption of factor VII ~ shortest half life)
– +/- Elevated PTT
– Low fibrinogen
– Elevated D-Dimer
– Low platelets
– MAHA (~50% of patients)
* Coagulation studies will be NORMAL in TTP – one way to differentiate from DIC.
Thrombotic Thrombocytopenia Purpura (TTP):
Due to a deficiency with ADAMTS13
Pentad (remember FAT RN) of TTP:
1) Fever
2) Anemia (microangiopathic hemolytic)
3) Thrombocytopenia
4) Renal disease
5) CNS disease (encephalopathy)
Other causes of MAHA (not a complete list):
– DIC
– HELLP
– Antiphospholipid syndrome
– Malignant hypertension
– Vasculitis
– Scleroderma renal crisis
– Many more…
Hereditary Hemochromatosis:
Triad:
Diabetes
Cirrhosis
Skin bronzing


Associated conditions:
– CPPD
– Arthropathy
– Hypogonadism
– Heart disease
– Destructive arthritis
Treatment:
Phlebotomy
*Caution patients about eating raw seafood or undercooked pork – increased incidence of Vibro vulnificus and Yersinia entercolitica in iron overload
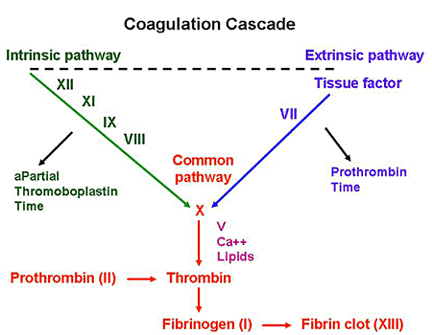
von Willebrand Disease (vWD):
– MOST COMMON inherited bleeding disorder
– Symptoms similar to platelet disorders: nosebleeds, easy bruising, bleeding gums, and post-surgical bleeding; GYN bleeding is especially common.
Underlying problem:
– vWF protects factor VIII from degradation
Lab abnormalities:
– prolonged aPTT (see below) – due to degradated factor VIII
Treatment:
– DDAVP (desmopressin) – causes preformed stores of vWF and factor VIII to be released from endothelial cells
Hereditary Spherocytosis:
– Due to mutations causing deficiencies/dysfunction in erythrocyte membrane proteins – reducing surface-to-volume ratio
– Patient’s are at an increased risk of pigmented (bilirubin) gallstones
Lab abnormalities:
– Spherocytes on blood smear
– Varying degrees of anemia/reticulocytosis/bilirubin elevation
– ↑ MCHC reflecting membrane loss – CHARACTERISTIC
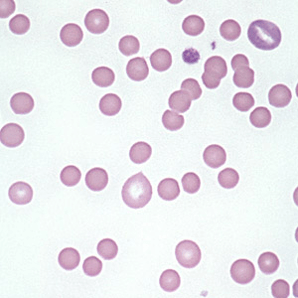
– see spherocytes on peripheral smear
– Osmotic fragility test can aid in the diagnosis when it is not clear (i.e. no family history)
Treatment options:
– Folate supplementation
– Splenomegaly (reduces hemolysis) ~ reserved for severe form; ensure vaccination against encapsulated organisms S. pneumoniae, H. influenza, N. meningitidis
Polycythemia vera (PV):
– PV is a disorder of myeloid/erythroid stem cell that causes erythropoietin-independent proliferation of erythrocytes
– Activating mutation of JAK2 (JAK2 V617F) is present in ~95% of PV
First step in making the diagnosis of PV:
– Exclude secondary causes of elevated Hct/red cell mass: chronic hypoxia and excess erythropoietin
Clues to the diagnosis of PV:
– Itching after a shower (aquagenic pruritus)
– Painful/red palms/soles (erythromelagia)
– Splenomegaly
Treatment:
– Phlebotomy (goal Hct <45%)
– +/- Myelosuppression (hydroxyurea)
– Aspirin
*Look out for a question that introduces acute leukemia in a patient with a history of PV
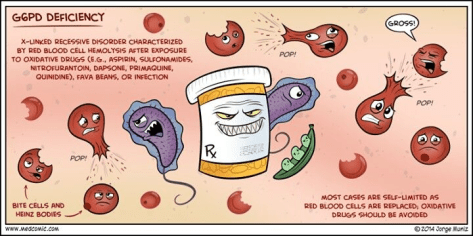
Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD):
– X-linked – therefore men are primarily affected
– G6PD is important for cells to counterbalance oxidative stress
Known triggers (not a full list):
– Medications: Sulfonamides, Nitrofurantonin, Anti-malarials, Rasburicase
– Naphthalene in mothballs
– Fava beans
– Infection
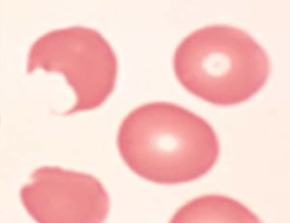
Peripheral smear:
Bite cells: membrane defect that appears as a semicircular “bite” has been taken out of an erythrocytes – caused by removal of denatured hemoglobin by macrophages in the spleen
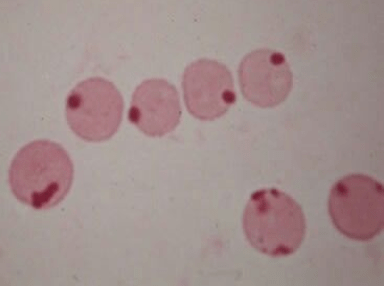
Heinz bodies: denatured oxidized hemoglobin visualized as intranuclear inclusions
Antiphospholipid Antibody Syndrome (APLS):
– Need ONE LAB criteria (confirmed 12 weeks apart) and ONE CLINICAL criteria
Lab Criteria:
– B2 Glycoprotein
– Anti-Cardiolipin antibody
– Lupus anticoagulant (measured as DRVVT that does not correct with mixing study)
*Positivity of all three lab tests is associated with the highest risk of thrombosis and pregnancy loss.
Clinical Criteria:
– Any thrombosis (venous or arterial)
– Fetal loss/miscarriage
Clinical Features of APLS:
– Prolonged PTT (50%)
– Livedo reticularis (20%)
– Cardiac valvular disease ~ MR
– DVT (32%)
– Stroke (13%)
– Hemolytic anemia (7%)
Paroxysmal Nocturnal Hemoglobinuria (PNH):
– PNH is an acquired disease – results in cells (RBCs, WBCs, platelets) lacking normally attached surface proteins
– CD55/CD59 are responsible for inactivating complement on RBC surface; lacking this protein, therefore, results in more complement-mediated lysis
Why does it occur at night?
– Complement-mediated lysis occurs more readily at lower pH levels and PCO2 levels rise at night – so patients report AM hematuria
Other associated conditions (besides hemolysis)
– Developing thrombosis (at unusual sites) – not fully explained; consider PNH in patients with both venous/arterial clots – Budd-Chiari syndrome may be a clue!
Treatment:
– Eculizumab – anti-CD5 monoclonal antibody that ceases hemolysis by inhibiting complement
Hairy Cell Leukemia (HCL):
Two clues to help make the diagnosis:
– Photo showing thread-like projections emanating from the cell surface (i.e. “hairy” appearing cells)
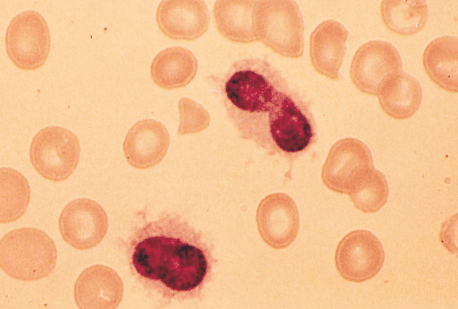
– Bone marrow biopsy resulting in a “dry tap” – occurs when marrow is difficult to extract due to firbrosis (seen in some cases of HCL)
Waldenström Macroglobulinemia:
– Key is to know that Waldenström’s overproduces the IgM paraprotein, but myeloma rarely does – it usually overproduces the IgG
Diagnosis:
– Clinical
– IgM monoclonal gammopathy
– Marrow biospy showing >10% lymphoplasmacytic cells
– Flow cytometry
Treatment:
– Plasmapheresis for hyper-viscosity
– Rituximab
