Etiologies of Chronic Pancreatitis (progressive inflammatory changes in the pancreas that result in permanent structural damage and histologic fibrosis)
–Alcohol abuse (45 %) as well as cigarette use
-Recurrent acute pancreatitis
–Genetic (eg: CFTR, SPINK mutations)
-Chronic ductal obstruction
-Systemic diseases (eg: SLE, hyperparathyroidism, hypertriglyceridemia)
-Idiopathic
–AUTOIMMUNE–Can be Type 1 (part of IgG4 disease) vs. Type 2 (idiopathic duct-centric pancreatitis without systemic involvement or IgG4 +)
Clinical manifestations of chronic pancreatitis
-Can be ASYMPTOMATIC
–Epigastric abdominal pain most common symptom however
–Pancreatic insufficiency (only after 90 % of pancreatic function lost) and manifests as steatorrhea and glucose intolerance/diabetes
-Remember that chronic pancreatitis puts you at increased risk for PANCREATIC CANCER
Lab/Imaging
-Amylase/Lipase usually NORMAL so not as helpful
–72 hour quantitative fecal fat (steatorrhea alone is non-specific!)
–Fecal elastase has high sensitivity and specificity for chronic pancreatitis
-KUB can show calcifications hinting towards chronic pancreatitis and MRCP/ultrasound can show pancreatic duct obstructions, dilations, strictures or fluid collections
Treatment
-Alcohol and smoking cessation!
-Creon supplementation, may also need fat-soluble (A,D,K,E) supplementation
-Analgesics for abdominal pain which is extremely hard to control. Minimize opioids but may be necessary for refractory pain.
-Specialized approaches include celiac nerve blocks, endoscopic surgery and surgical resection
IgG4 related disease
–Inflammatory and Fibrotic systemic condition where organs have tumefactive (swelling), IgG4 positive lymphoplasmacytic infiltrate with often elevated IgG4 serum levels but not always
Clinical manifestations of IgG4 related disease
–Pancreatic involvement (manifests as pancreatic mass which can mimic CANCER, recurrent pancreatitis, strictures etc.)
–Biliary involvement (biliary strictures leading to obstructive jaundice as well as sclerosing cholangitis)
-ANY organ can be affected (eg: thyroiditis, interstitial nephritis, salivary involvement) Diagnosis and Treatment
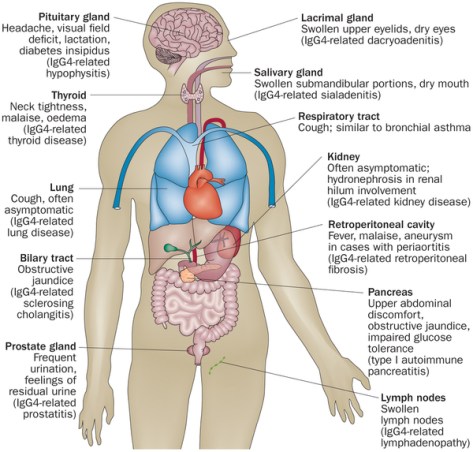
http://www.nature.com/nrrheum/journal/v10/n3/full/nrrheum.2013.183.html
Diagnosis
-Diagnosis is made with the HISORt criteria (need at least one criteria)-see below

–Treatment is with STEROIDS and may need immunomodulating therapy depending on clinical course.
Further reading