Thanks to Cameron and Adam for presenting the case of a middle aged man with no significant PMH who presented with diffuse myalgias and chronic progressive proximal muscle weakness, found to have a CK >12k and EMG findings concerning for an inflammatory myopathy, awaiting muscle bx for diagnosis.
Clinical Pearls
- Rhabdomyolysis literally means dissolution of skeletal muscle and has a broad differential outside of the typical traumatic or exertional processes associated with it see below).
- The four main inflammatory myopathies are dermatomyositis, polymyositis, inclusion body myositis, and necrotizing autoimmune myositis.
- Polymyositis is rare and a diagnosis of exclusion after the other three main inflammatory myopathies have been investigated.
- Overall, the prognosis of inflammatory myopathies is good with appropriate treatment. The exception is inclusion body myositis which is a progressive disorder without any effective therapy.
- Pigment nephropathy can occur with rhabdo regardless of the underlying etiology especially in patients with CK >5000. Aggressive IV hydration to lower CK levels is important to reduce the risk of kidney injury.
Rhabdomyolysis:
DDx:
-
Traumatic
-
Crush injuries, surgery, prolonged compression from immobility or coma
-
-
Non-traumatic
-
Exertional:
-
Normal muscle: strenuous exercise, heat stroke, seizures, hyperkinetic states
-
Abnormal muscle: metabolic myopathies, mitochondrial myopathies, malignant hyperthermia, NMS
-
-
Non-exertional
-
Alcoholism
-
Drugs and toxins: lipid-lowering drugs (fibrates, statins), alcohol, heroin, cocaine, meth, colchicine
-
Infections: influenza, coxsackie, EBV, HIV, legionella
-
Electrolyte abnormalities: hypokalemia, hypophosphatemia, hypocalcemia
-
Endocrinopathies: DKA, HHS, hypothyroidism, vitamin D deficiency
-
Inflammatory myopathies (rare)
-
Paraneoplastic
-
Miscellaneous
-
-
Inflammatory myopathies
Largest group of potentially treatable myopathies in children and adults.
-
Four subtypes: distinguishing which process is important because each subtype has a different prognosis and response to therapy
-
DM
-
Anti-Mi-2, anti-MDA-5, anti-TIF-1, anti-NXP-2
-
-
PM
-
Rare, often misdiagnosed
-
Dx of exclusion
-
-
Necrotizing autoimmune myositis
-
More common than PM
-
Occurs alone or after viral infections or in association with cancer, CTD, or post-statin
-
Anti-SRP or anti-HMGCR
-
Highest CK level
-
-
Inclusion body myositis
-
Most common in people >50
-
7.9 cases/million in the US
-
Distal muscles impacted first
-
Facial muscles impacted
-
Muscle atrophy occurs earlier than in others
-
Extramuscular manifestations are uncommon
-
Dysphagia occurs in >50%
-
Muscle atrophy is common
-
Lowest CK level
-
-
-
Up to 30% of patients with DM or PM have a constellation of clinical findings termed “antisynthetase syndrome”
-
Acute disease onset
-
Constitutional symptoms (fever, weight loss)
-
Myositis
-
Raynaud’s
-
Mechanic’s hands
-
Non-erosive arthritis
-
ILD
-
Labs show antibodies to tRNA synthetase enzymes (anti-Jo-1)
-
-
Extramuscular manifestations
-
systemic symptoms
-
cardiac arrhythmias or ventricular dysfunction
-
pulmonary complications (ILD)
-

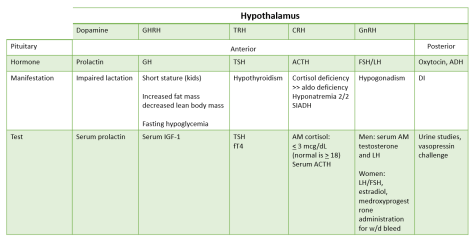
Table above adapted from this and this review article by NEJM.