Today we discussed a fascinating case of statin-related anti-HMGCR positive immune-mediated inflammatory myositis (also called necrotizing autoimmune myositis). The case highlighted the importance of a framework approach to diseases.
We first went over the framework for true muscle weakness, which can be anatomically divided as follows
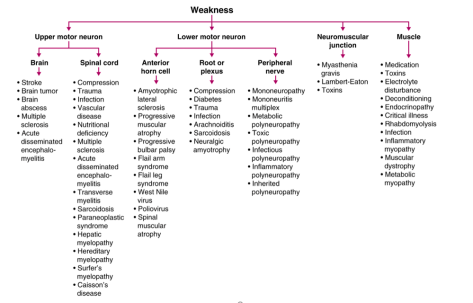
Source: Frameworks for Internal Medicine (Dr. Andre Mansoor from OHSU). Available on Amazon (highly recommended!)
To help us localize the lesion to a myopathy, we used the following framework to determine that it was likely a myopathy.

The differential for myopathy is broad, and generally is the same for an elevated CK and non-traumatic, non-exertional rhabdomyolysis. The causes can be divided as follows. If you like mnemonics, think Drug-REGIIME for the various categories.

Once we narrowed the differential to an inflammatory myopathy, we utilized the following chart that guided us to the probable conclusion that it was an immune-mediated necrotizing myopathy (also known as necrotizing autoimmune myopathy). This was confirmed by a highly positive anti-HMGCR antibody
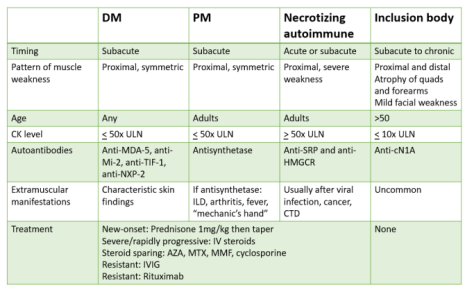
Adapted from a NEJM Article: https://www.nejm.org/doi/full/10.1056/NEJMra1402225
Clinical Pearls about INMN/NAM:
- Can occur in association with viral infections, malignancy, in patients with CTD such as scleroderma or in patients taking statins
- Can start acutely or sub-acutely with severe proximal muscle weakness and markedly elevated CK values of between 10-100k ULN (2K-20K)
- A rare diagnosis, but some experts believe that polymyositis is overdiagnosed and that INMN may actually be more common!
- Two antibodies are highly specific though not sensitive for the condition (anti-SRP and anti-HMGCR)
- The majority of anti-HMGCR positive cases are related to known exposure to a prescribed statin (~70%)
- Treatment is to discontinue the statin but most cases will require prompt immunosuppression
- Statin-induced muscular events are on a spectrum, and include the following:
- 1) Mildly elevated CK and myalgias
- 2) Rhabdomyolysis
- 3) Self-limited toxic myopathy
- 4) IMNM
- It is only IMNM that generally does not improve with merely cessation of the drug and it generally needs immunosuppression
- Statin-induced muscular events are on a spectrum, and include the following:
- Biopsy is required to make the diagnosis but the presence of the antibody will often result much more quickly and in our case, the patient started on immunosuppresion prior to the biopsy results because the clinical profile fit perfectly with this diagnosis