Today, we talked about the case of a middle-aged man from the Philippines who presented with a one year progressive pruritic rash involving the face, arms, and legs as well as a distal symmetric peripheral neuropathy, found to have lepromatous leprosy on skin biopsy!
Clinical Pearls
- Mycobacterium leprae and lepromatosis like to grow in cooler areas, so infection often manifests in the skin and the peripheral nerves.
- Transmission is likely via respiratory route, through broken skin, and by touching armadillos!
- Early recognition and treatment is important to prevent injury to peripheral nerves.
DDx for rash + neuropathy
- Lyme (usually cranial nerves, radiculopathy)
- Celiac
- Zoster (tends to be painful rather than pruritic and localized to a dermatome)
- WNV (flaccid paralysis)
- Sarcoid
- Amyloid
- Syphilis
- Leprosy
Our patient presented with a pruritic rash and largely a distal symmetric peripheral neuropathy. We generated the following Venn diagram in report to help us with the diagnosis:

Leprosy
- AKA Hansen’s disease
- Infection caused by mycobacterium leprae and mycobacterium lepromatosis, separate species that cause similar clinical disease. They are both obligate intracellular parasites.
- Involves the skin and peripheral nerves
- Early treatment is important to prevent involvement of the eyes, hands, and feet due to neuropathy. The neuropathy is often non-reversible.
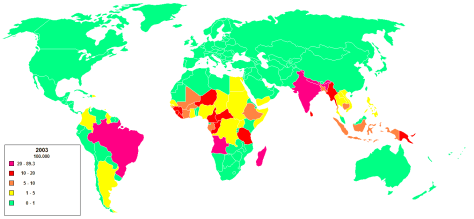
- 205 new cases detected in the US in 2010. 75% among immigrants (most commonly India, Brazil, Indonesia, Bangladesh, and Nigeria)
Transmission
- Unknown but probably respiratory route especially in lepromatous leprosy. Sometimes can transmit through broken skin. Also from armadillos.
- Most people do NOT develop disease after exposure. Risk factor for disease development include older age, genetic influences, and immunosuppression.
- Grows in cooler areas
Clinical presentation:
- Described in categories pertaining to how much bacillary burden of disease is present with tuberculoid being the least amount and lepromatous having the highest disease burden.
- Clinical features:
- Hypopigmented or reddish patches on the skin
- Typically involve the earlobes with nodular thickening and distributed symmetrically on the body in lepromatous leprosy.
- Diminished sensation or loss of sensation within skin patches
- Paresthesias of hands/feet
- Neuropathy occurs early in disease course
- Painless wounds or burns on the hands or feet
- Lumps or swelling on the earlobes or face
- Tender, enlarged peripheral nerves
- Hypopigmented or reddish patches on the skin
- Late findings in disease course:
- Weakness of the hands with claw fingers, foot drop, facial paralysis, lagophthalmos (can’t close eyes completely due to CN7 palsy), lack of eyebrows/eyelashes, collapsed nose, perforated nasal septum.
- Intermittent bacteremia can lead to focal lesions in various organs (liver, bone marrow, testicles and larynx)
Diagnosis:
- Consider it in patients with skin lesions and/or enlarged nerve(s) accompanied by sensory loss.
- No reliable blood or skin tests available.
- Usually clinical and skin biopsy
Treatment:
- Goal: Prevent and/or minimize injury to peripheral nerves!
- Often times it’s loss of sensation but later can progress to painful neuropathy
- Dapsone plus rifampin for tuberculoid leprosy. Clofazimine is added for lepromatous leprosy.
- Duration can be up to 24 months
- Treat neuritis with steroids for a prolonged course
- Make sure to screen for G6PD deficiency before prescribing dapsone
- Monitor liver function with rifampin
- Clofazimine (causes phototoxicity) is not available in US pharmacies and must be obtained from the NHDP.
Prognosis:
- May take a few years for skin lesions to resolve completely with treatment
- Very curable, low relapse rates, typically no drug resistance