
Today, we talked about the fascinating case of a middle-aged man presenting with subacute cough, night sweats, and 15 pound weight loss, found to have bilateral hilar LAD on CXR and CT concerning for pulmonary sarcoidosis. While awaiting LN biopsy, he developed L sided Bell’s palsy with MRI showing inflammation of CN5 and CN7 as well as nodular dural thickening of the trigeminal cave concerning for neurosarcoidosis. LN biopsy showed non-caseating granulomas and he was subsequently started on high dose steroids for neurosarcoidosis.
Clinical Pearls
- Sarcoidosis is a multisystem granulomatous disorder of unknown etiology.
- The most common clinical presentation of sarcoidosis is pulmonary related.
- In asymptomatic individuals with incidental hilar LAD, there is no indication for LN bx to diagnose sarcoidosis because 2/3 of cases resolve spontaneously without treatment. Close follow up is needed to ensure that patients do not develop symptoms.
- Similarly, Lofgren syndrome (arthritis, erythema nodosum, hilar LAD, and fevers) and Heerfordt’s syndrome (uveoparotid fever) are clinically consistent with sarcoidosis and there is no indication for LN biopsy.
DDx for unilateral facial droop
- CNS lesion
- Especially if forehead is spared. However, keep in mind that a stroke involving the region of pons which houses the nucleus of CN7 would mimic Bell’s palsy!
- PNS lesion
- Infections
- Zoster
- HIV
- Lyme
- Syphilis
- Malignancy
- Parotid tumors
- Acoustic neuroma/schwannoma
- Lymphoma or other mass compressing CN7
- Infiltrative/autoimmune
- Sjogren’s syndrome
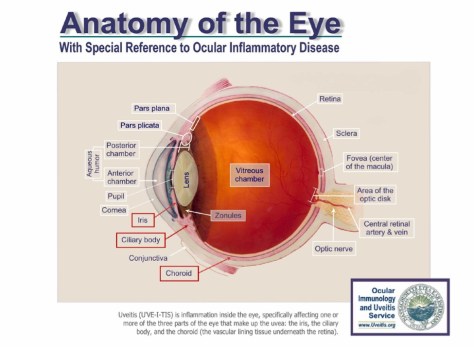
- Sarcoidosis
- Infections
Sarcoidosis
- Multisystem granulomatous disorder of unknown etiology
- 3-4 x more common in blacks
- Overall prevalence is 10-20 per 100,000 people
- Pathophys
- Aberrant formation/accumulation of non-caseating granulomas
- Clinical presentation
- Age of onset is 20-60 years
- Often discovered incidentally on a CXR
- Most common organ involved is the lung (cough, SOB, CP) with ILD being the most common type of lung involvement. 30% of patients present with extrathoracic manifestations.
- Fatigue, malaise, fever, and weight loss are common
- Lofgren’s syndrome: arthritis, erythema nodosum, b/l hilar LAD
- Heerfordt’s syndrome: parotid gland enlargement, facial palsy, fever, anterior uveitits
- Diagnosis
- No biopsy needed in the following
- Asymptomatic with hilar LAD
- Lofgren’s syndrome
- Heerfordt’s syndrome
- Otherwise, biopsy the easiest site to access. Keep in mind that erythema nodosum does not have granulomas and would not help in diagnosing sarcoidosis.
- Lab abnormalities that may be present:
- Anemia
- Leukopenia, eosinophilia, thrombocytopenia
- ESR and CRP may be elevated
- Hypercalciuria is more common than hypercalcemia
- Moderate elevation in ALP suggests diffuse granulomatous hepatic involvement
- Hypergammaglobulinemia and a positive RF (not usually part of routine work up)
- ACE levels are elevated in 75% of untreated patients with sarcoid but has poor sensitivity and insufficient specificity (10% false positive rate with cocci, DM2, TB, hyperthyroidism, lung cancer, pneumoconiosis, etc.)
- No biopsy needed in the following
- Organs that may be impacted in sarcoid
- Pulmonary: occurs in over 90% of patients. Bilateral hilar LAD as well parenchymal disease. Can present with a restrictive spirometry pattern due to underlying fibrosis, pulmonary HTN
- Cutaneous: can be disfiguring can be macules, papules, plaques and erythema nodosum
- Liver/Spleen: a high alkaline phosphatase level suggests granulomatous liver disease
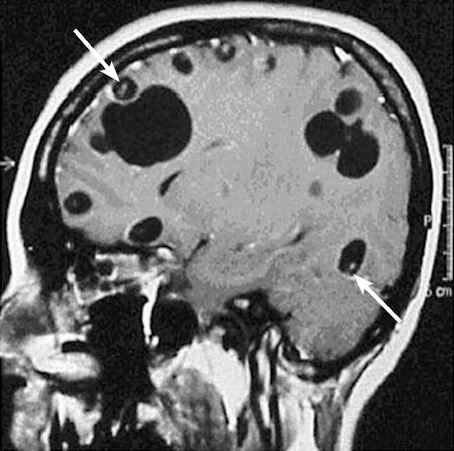
- Neurologic: Noted in 5% of cases. MRI with contrast can help with diagnosis. Complications:
- Cranial-nerve palsies (20-50%) → most common
- Headache
- Ataxia
- Encephalopathy
- Granulomatous meningitis
- Weakness
- Seizures
- Neuroendocrine dysfunction
- Optho: anterior uveitis most common manifestation
- Cardiac: Typically cardiomyopathy, can also see arrhythmias (tachy or brady).
- Renal: Can have hypercaliuria (more common than hypercalcemia) and renal calculi
- Bone: chronic arthritis and cysts resembling rheumatoid, and diffuse granulomatous myositis.
- Treatment/monitoring:
- Asymptomatic? Follow up outpatient q3-4 months and annually thereafter to monitor for development of symptoms.
- Symptomatic? Start steroids, re-evaluate q1-2 months
- Refer to this NEJM article for organ specific treatments.
- Chronic complications of pulmonary sarcoid
- VTE is more common than the average patient population
- Chronic pulmonary aspergillosis
- Pulmonary HTN due to advanced fibrosis
- Prognosis
- Up to 80% of patients with hilar LAD spontaneously improve on their own!
- With more symptomatic disease or more extrapulmonary manifestations, prognosis declines to less than 30% remission.
- Overall mortality is <5%






 Source: NEJM, Grepmed
Source: NEJM, Grepmed
