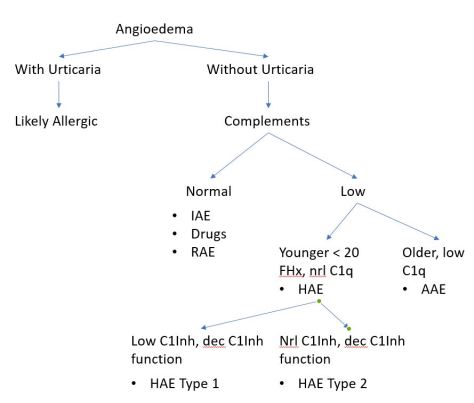
Today our Doctor-in -Training, Emma, presented a case of a middle age woman with no significant medical or family history coming in with a month of fatigue, menorrhagia, and gum bleeding. She was incidentally found to be pan-cytopenic. Bone marrow biopsy revealed hypocellularity with no abnormal morphologies, which is consistent with aplastic anemia!
Aplastic anemia
- Epidemiology
- Rare, 2 per million per year, higher incidence amongst Asians, can occur in all ages but most of the time in the first 1st-4th decades.
- Pathophysiology
- Big picture: Think of it this way. Aplastic anemia is similar to a factory not working due to not enough workers (stem cells). Compare and contrast this with MDS, which is basically a loss of quality control (mutation in the stem cells) leading to bad products that get worse over time (leading eventually to pancytopenia, leukemia).

- Causes
- Primary (rare), most of the time acquired
- Infectious: CMV, Parvo, EBV, HIV, hepatitis, TB
- Toxins: Benzene, glue vapors, solvents, pesticides
- Autoimmune: SLE, eosinophilic fasciitis, graft v host,
- Other: Pregnancy, paroxysmal nocturnal hemoglobinuria, thymoma, irradiation
- Medications: NSAIDS, Bactrim, Lasix, Gold, Allopurinol, penicillamine, phenothiazines, AEDS, anti-psychotics
- Association
- PNH: CD55 & CD59 abnormalities can be sen in up to 50% of patients with aplastic anemia!
- Presentation
- S/S of anemia
- S/S of thrombocytopenia (menorrhagia, petechiae)
- Recurrent infections
- Diagnosis
- Bone marrow biopsy most definitive, helps distinguish AA from other causes of pancytopenia
- BM finding
- Hypocellular, marrow space mostly fat cells and marrow stroma.
- Remaining cells are morphologically normal
- No fibrosis, no e/o malignancy
- Severe AA criteria
- BM cellularity < 25 + 2 of ANC < 500, plt < 20k, retic < 20k
- Very Severe AA criteria
- ANC < 200
- Mgx: Severe AA require treatment
- Under age 20: Early allogeneic HCT as soon as possible. If HCT not possible, immunosuppressives
- 20 – 50: HCT also recommended if no contraindications, immunosuppressives (Anti-thymocyte globulin, cyclosporine for example) + eltrombopag (thrombopoietin agonist) otherwise.
- > 50:
- Transplantable: Early hematopoietic cell transplantation
- Older, not candidates for HCT: Immunosuppressive, less intense preferred as age increases.
- Treat other underlying/reversible causes in all cases.
- Causes
- Big picture: Think of it this way. Aplastic anemia is similar to a factory not working due to not enough workers (stem cells). Compare and contrast this with MDS, which is basically a loss of quality control (mutation in the stem cells) leading to bad products that get worse over time (leading eventually to pancytopenia, leukemia).
Fun Fact: Blood transfusion can lead to falsely elevated iron studies within the first 24-36 hours.